醫(yī)療器械出口到馬來西亞需要滿足一些特定的認(rèn)證和要求。以下是醫(yī)療器械出口馬來西亞所需的認(rèn)證和注意事項:
一、醫(yī)療器械立法及主管
馬來西亞的醫(yī)療器械管理法是《醫(yī)療器械管理法(2012)》,該法規(guī)的基本框架比較接近美國的醫(yī)療器械管理法規(guī)。醫(yī)療器械的管理部門包括衛(wèi)生部醫(yī)療器械管理局(Medical Device Authority,MDA)和科學(xué)技術(shù)部原子能許可證局。衛(wèi)生部醫(yī)療器械管理局負(fù)責(zé)管理除放射性醫(yī)療器械和二手醫(yī)療器械外的所有醫(yī)療器械,而原子能許可證局負(fù)責(zé)管理放射性醫(yī)療器械和二手醫(yī)療器械。
二、馬來西亞進(jìn)口醫(yī)療器械市場準(zhǔn)入流程
(1)馬來西亞醫(yī)療器械的定義及分類
醫(yī)療器械被定義為具備以下用途的任意或聯(lián)合使用的儀器、器械、器具、機(jī)器、植入物、體外試劑或校準(zhǔn)器、軟件、材料等。它們可以用于診斷、預(yù)防、監(jiān)測、治療、減輕疾病及損傷,對解剖學(xué)或生理過程的研究,支持或維持生命,對器械的消毒,以及通過對取自人體的標(biāo)本進(jìn)行體外檢查,為醫(yī)療或診斷目的提供信息。然而,如果通過藥物、免疫等過程發(fā)揮這些功能,則不能被定義為醫(yī)療器械。根據(jù)馬來西亞醫(yī)療器械管理法規(guī),醫(yī)療器械按風(fēng)險從低到高分為A類、B類、C類和D類四類。其中,A類醫(yī)療器械風(fēng)險最低,B類和C類居中,D類風(fēng)險最高。
A類產(chǎn)品:低風(fēng)險,如細(xì)胞計數(shù)儀。
B類產(chǎn)品:中低風(fēng)險,如尿液分析儀。
C類產(chǎn)品:中高風(fēng)險,如心電圖儀。
D類產(chǎn)品:高風(fēng)險,如血液透析機(jī)。
(2)體外診斷試劑的分類及注冊
體外診斷試劑(in-vitro diagnostic,IVD)是指僅或主要用來為人體提供診斷、監(jiān)測信息而單獨或組合使用的裝置,包括試劑、校準(zhǔn)器、樣品容器等。根據(jù)醫(yī)療器械管理法,IVD產(chǎn)品根據(jù)對個人及公眾健康風(fēng)險影響程度的高低分為A、B、C、D四類。各類別的風(fēng)險程度和產(chǎn)品舉例如下:

對于IVD的注冊,需要遵循醫(yī)療器械管理法中的專門適用準(zhǔn)則,并按照相應(yīng)的步驟進(jìn)行注冊。

三、馬來西亞進(jìn)口醫(yī)療器械注冊流程
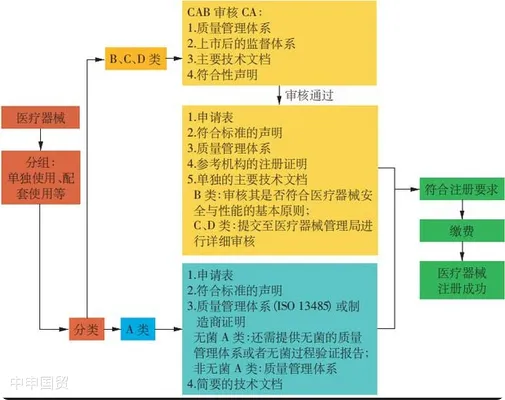
(1)A類產(chǎn)品
對于A類產(chǎn)品,當(dāng)?shù)氐尼t(yī)療器械授權(quán)代表(Authorized Representative,AR)可以向衛(wèi)生部醫(yī)療器械管理局申請注冊,無需MDA授權(quán)的合格評定機(jī)構(gòu)(Conformity Assessment Body,CAB)批準(zhǔn)。申請人需要提交制造商的ISO 13485證書、測試報告以及標(biāo)簽等文件。
(2)B、C、D類產(chǎn)品
對于B、C、D類產(chǎn)品,當(dāng)?shù)氐尼t(yī)療器械授權(quán)代表需要提交技術(shù)報告,并由合格評定機(jī)構(gòu)進(jìn)行技術(shù)文件審查。已經(jīng)在參考國(如澳大利亞、加拿大、歐盟、日本、美國)獲得批準(zhǔn)和銷售的醫(yī)療器械可以通過簡化程序進(jìn)行審核。在審核過程中,需要向合格評定機(jī)構(gòu)提交ISO證書、CE證書等文件。審核通過后,合格評定機(jī)構(gòu)將頒發(fā)證書。最終的器械注冊文件,包括通用提交檔案模板(Common Submission Dossier Template,CSDT)、合格評定機(jī)構(gòu)證書和申請文件,將以電子方式在線提交給衛(wèi)生部醫(yī)療器械管理局進(jìn)行審查和最終批準(zhǔn)。合格評定機(jī)構(gòu)的證書和器械注冊證書均應(yīng)每5年更新一次。
四、注意事項
在醫(yī)療器械出口到馬來西亞的過程中,需要注意以下事項:
(1)目前馬來西亞注冊已不再需要自由銷售證書(Free Sales Certificate,F(xiàn)SC)。
(2)必須在馬來西亞指定的當(dāng)?shù)卮硖幪峤蛔陨暾垺?br>(3)每個產(chǎn)品只能有一個產(chǎn)品許可證持有人(即持證人)。
(4)產(chǎn)品許可證可以轉(zhuǎn)讓給其他持有人。
(5)所有制造商需要獲得ISO 13485認(rèn)證,作為申請注冊的必備條件。
總結(jié)而言,醫(yī)療器械出口到馬來西亞需要滿足醫(yī)療器械管理法規(guī)定的注冊要求。具體流程包括分類確定、技術(shù)文件審查和提交、合格評定機(jī)構(gòu)認(rèn)證以及最終的在線注冊審批。在申請注冊之前,應(yīng)仔細(xì)了解相關(guān)法規(guī)和要求,并確保符合馬來西亞的醫(yī)療器械出口標(biāo)準(zhǔn)。
標(biāo)簽: 醫(yī)療器械進(jìn)出口





? 2025. All Rights Reserved. 滬ICP備2023007705號-2  滬公網(wǎng)安備31011502009912號
滬公網(wǎng)安備31011502009912號